Familial amyloid polyneuropathy, also called transthyretin-related hereditary amyloidosis, transthyretin amyloidosis abbreviated also as ATTR (hereditary form), or Corino de Andrade’s disease is a severe neurodegenerative genetic disease.
It is rare, with an estimated 8,000 people affected worldwide.
Foci of familial amyloid polyneuropathy
The first cases were detected in northern Portugal in the 16th century. It was called “the foot disease”, as the first symptom of the disease was the dragging of the feet. Currently, the main foci of the disease are in Portugal, Sweden and Japan. The prevalence of cases in Spain is highest in the Balearic Islands. In fact, Majorca is the fifth world focus of the disease. The prevalent mutation in the Mallorcan population is known as Val30Met.
Cause and symptoms of familial amyloid polyneuropathy
Familial amyloid polyneuropathy is caused by a mutation in the gene encoding the Transthyretin (TTR) protein. The mutation in the gene causes deficient synthesis of the protein. This abnormal protein, which is known as amyloid substance, is produced in the liver and from there it is transported to different parts of our body. Eventually, it is deposited in the nervous system and other parts of the body such as the intestines, kidneys or heart. Over time, this continuous deposition leads to peripheral, sensory-motor and autonomic neuropathy.
Andrade’s disease is irreversible, progressive and fatal. It mainly affects adults from the third decade of life onwards and the first symptoms are paraesthesia (abnormal sensations of numbness, tingling, tingling, numbness), pain or trophic (non-healing) lesions of the feet, gastrointestinal disorders and weight loss. In more advanced stages, more pronounced sensory loss involves pain and variations in wind chill. In severe stages, complete motor loss occurs.
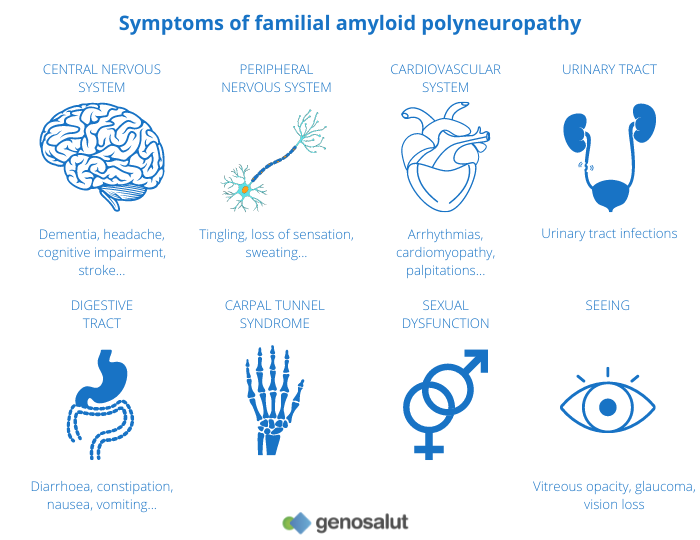
Diagnosis and treatment of familial amyloid polyneuropathy
Diagnosis is complex, as the symptoms appear differently in each case. Clinically, it is very difficult to distinguish it from other peripheral neuropathies and it can sometimes take months or even years before it is correctly diagnosed. Life expectancy is variable and depends largely on early diagnosis. If left untreated, it can lead to death within 10 years.
Until a few years ago, treatment options were limited to symptom management or liver transplantation. However, in the last decade, two drugs have come on the market that help slow the progression of the disease. Tafamidis (Pfizer) acts as a protein stabiliser for patients diagnosed early. It avoids the use of immunosuppressants, is less invasive, and is not contraindicated for people over 70. The other drug is Partisiran (Alnylam), an RNAi drug that works by silencing the gene involved in the formation of the defective protein that accumulates in various organs. On 3 April this year, the Directorate General for the Common Portfolio of NHS Services and Pharmacy included ONPATTRO® (trade name of Patisiran) in the NHS pharmaceutical benefit for the treatment of hereditary amyloidosis due to transthyretin (AhTTR) with polyneuropathy in stages 1 or 2.
At Genosalut we analyse the Val30Met mutation as well as other mutations associated with Andrade disease.
Si te gusta nuestro blog, suscríbete a nuestro newsletter

