Die familiäre Amyloidpolyneuropathie Typ I, die Andrade-Krankheit oder die hereditäre transthyretinbezogene Amyloidose ist eine schwere genetisch bedingte neurodegenerative Erkrankung.
Sie ist selten, weltweit sind schätzungsweise 8.000 Menschen betroffen.
Herde der Andrade-Krankheit
Die ersten Fälle wurden im 16. Jahrhundert in Nordportugal entdeckt. Sie wurde „die Fußkrankheit“ genannt, da das erste Symptom der Krankheit das Schleifen der Füße war. Gegenwärtig befinden sich die Hauptzentren der Krankheit in Portugal, Schweden und Japan. Die Prävalenz der Fälle in Spanien ist auf den Balearen am höchsten. Tatsächlich ist Mallorca der fünfte weltweite Schwerpunkt der Krankheit. Die in der mallorquinischen Bevölkerung vorherrschende Mutation ist als Val30Met bekannt.
Ursache und Symptome der Andrade-Krankheit
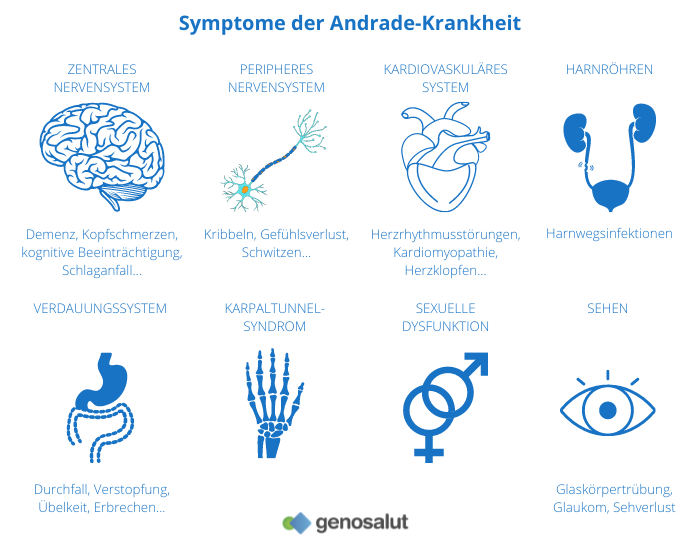
Die Andrade-Krankheit wird durch eine Mutation in dem Gen verursacht, das für das Protein Transthyretin (TTR) kodiert. Die Mutation des Gens verursacht eine mangelhafte Synthese des Proteins. Dieses abnorme Protein, das als Amyloid bekannt ist, wird in der Leber produziert und von dort in verschiedene Teile unseres Körpers transportiert. Schließlich lagert es sich im Nervensystem und in anderen Körperteilen wie dem Darm, den Nieren oder dem Herzen ab. Mit der Zeit führt diese kontinuierliche Ablagerung zu einer peripheren, sensomotorischen und autonomen Neuropathie.
Die Andrade-Krankheit ist irreversibel, fortschreitend und tödlich. Sie betrifft vor allem Erwachsene ab dem dritten Lebensjahrzehnt, und die ersten Symptome sind Parästhesien (abnormale Empfindungen von Taubheit, Kribbeln, Kribbeln, Taubheit), Schmerzen oder trophische (nicht heilende) Läsionen der Füße, gastrointestinale Störungen und Gewichtsverlust. In fortgeschrittenen Stadien ist der Verlust der Sinneswahrnehmungen ausgeprägter und geht mit Schmerzen und Schwankungen der Windkälte einher. In schweren Stadien kommt es zu einem vollständigen Verlust der Motorik.
Diagnose und Behandlung der Andrade-Krankheit
Die Diagnose ist komplex, da die Symptome in jedem Fall anders aussehen. Klinisch ist es sehr schwierig, sie von anderen peripheren Neuropathien zu unterscheiden, und es kann manchmal Monate oder sogar Jahre dauern, bis sie richtig diagnostiziert wird. Die Lebenserwartung ist unterschiedlich und hängt weitgehend von einer frühzeitigen Diagnose ab. Bleibt sie unbehandelt, kann sie innerhalb von 10 Jahren zum Tod führen.
Bis vor einigen Jahren waren die Behandlungsmöglichkeiten auf die Behandlung der Symptome oder eine Lebertransplantation beschränkt. In den letzten zehn Jahren sind jedoch zwei Medikamente auf den Markt gekommen, die das Fortschreiten der Krankheit verlangsamen können. Tafamidis (Pfizer) wirkt als Eiweißstabilisator für Patienten, bei denen eine frühe Diagnose gestellt wird. Sie macht den Einsatz von Immunsuppressiva überflüssig, ist weniger invasiv und ist für Menschen über 70 nicht kontraindiziert. Das andere Medikament ist Partisiran (Alnylam), ein RNAi-Medikament, das das Gen, das an der Bildung des defekten Proteins beteiligt ist, das sich in verschiedenen Organen ansammelt, zum Schweigen bringt. Am 3. April dieses Jahres hat die Generaldirektion für das gemeinsame Portfolio der NHS-Dienste und der Pharmazie ONPATTRO® (Handelsname Patisiran) in den pharmazeutischen Nutzen des NHS für die Behandlung der hereditären Amyloidose aufgrund von Transthyretin (AhTTR) mit Polyneuropathie im Stadium 1 oder 2 aufgenommen.
Bei Genosalut analysieren wir die Val30Met-Mutation sowie andere mit der Andrade-Krankheit assoziierte Mutationen.
Wenn Sie unseren Blog mögen, abonnieren Sie unseren Newsletter

