Què és la seqüenciació del genoma complet?
És una tècnica de seqüenciació massiva (NGS, next generation sequencing) que permet seqüenciar, de forma paral·lela, milions de fragments d’ADN, que cobreixen tot el genoma (WGS, whole genome sequencing)
Seqüenciació massiva: anàlisi del genoma
La seqüenciació del genoma complet (WGS) aborda (gairebé) tot l’ADN de la persona, tant les regions codificants (gens) com no codificants, i és capaç d’identificar la majoria dels canvis a l’ADN.
Aquesta eina, al principi exclusiva del camp de la investigació, ja porta gairebé una dècada introduïda a la pràctica clínica. A més, s’espera que en el futur adquireixi un paper més rellevant dins de l’àmbit sanitari gràcies al descens ràpid dels costos de seqüenciació, la capacitat de produir grans volums de dades amb els seqüenciadors actuals i l’avenç del coneixement de les variants genètiques.
En el futur de la medicina personalitzada, les dades de la seqüència del genoma complet poden ser una eina important per guiar la prevenció, la detecció i la teràpia dels pacients.
Avantatges i desavantatges de l'anàlisi del genoma complet
Avantatges de la seqüenciació del genoma complet
Els avantatges d’aquesta tècnica són:
- Proporciona una visió d’alta resolució, base per base, del genoma.
- Captura tant les variants grans com les petites que podrien passar desapercebudes amb enfocaments específics.
- Detecta variants d’un sol nucleòtid.
- Detecta insercions i delecions (com la deleció causant de l’atròfia muscular espinal).
- Detecta canvis al nombre de còpies.
- Detecta grans variants estructurals.
- Detecta translocacions equilibrades.
- Detecta repeticions, per exemple les comunes en diversos tipus de malalties neuromusculars. Per exemple, aquelles conegudes com a malalties “poliglutamíniques” que inclouen entre altres, la malaltia de Kennedy o atròfia muscular progressiva espinobulbar, la malaltia de Huntington, diverses atàxies espinocerebeloses i l’atròfia dentatorubro-pálidoluisiana.
- El significat de variants la influència sobre la salut de les quals es desconeix actualment es pot conèixer en el futur. Com que les dades es poden conservar, la persona pot ser informada en aquest cas.
- Proporciona tota la informació farmacogenètica.
Desavantatges de la seqüenciació del genoma complet
La dificultat més gran associada a la seqüenciació del genoma complet és l’enorme massa d’informació que proporciona, que ha de ser analitzada i avaluada per determinar el que és important o el que no ho és.
Tot i que els coneixements en genòmica són cada vegada més grans, les funcions de molts gens són encara indeterminades i es desconeix el paper de moltes variants (no se sap si són benignes o patògenes). Tot i això, gràcies a nous descobriments, pot ser una informació valuosa en el futur.
Aplicacions clíniques de l'anàlisi del genoma complet
L'anàlisi del genoma complet abasta múltiples aspectes de la salut:
- Identificar malalties monogèniques (com la talassèmia).
- Identificació de la possible causa en malalties rares.
- Identificar una predisposició a desenvolupar una malaltia poligènica (com la diabetis mellitus tipus 2).
- Identificar els portadors genètics de malalties recessives, com la fibrosi quística.
La importància d'una bona interpretació de les dades
La seqüenciació de l’exoma permet la identificació de moltes variants més (mutacions puntuals, insercions i delecions) que altres tècniques, encara que es desconegui el significat de part d’aquesta informació. Atès que no tots els canvis genètics afecten la salut, de vegades és difícil saber si les variants identificades estan implicades en la salut de la persona o malaltia d’interès. De vegades, una variant identificada s’associa amb un trastorn genètic diferent que encara no s’ha diagnosticat (el que s’anomena troballes incidentals o secundàries).
Per això, és imprescindible comptar amb un equip de professionals experts capaços d’interpretar les dades i donar resposta a les preguntes dels metges i els pacients.
Quines variants genètiques pot detectar la seqüenciació del genoma?
Les aplicacions de la seqüenciació del genoma complet (WGS, whole genome sequencing) han anat augmentant des dels inicis. Si bé al principi s’usava principalment per a la detecció de canvis en un sol nucleòtid (SNV, single nucleotide variant o SNP, single nucleotide polymorphism) o mutacions puntuals, actualment amb aquesta tècnica ja es poden detectar un altre tipus d’anomalies genètiques entre les que es troben:
- Petites variacions en el nombre de còpies com ara petites insercions i delecions
- Grans variacions en el nombre de còpies o CNVs (copy number variants)
- Repeticions curtes en tàndem (STR, short tandem repeat)
- Seqüències d’homozigositat (ROH, runs of homozygosity)
- Mutacions presents en un percentatge molt petit de cèl·lules (mosaïcismes i/o contaminacions)
- Reordenaments cromosòmics més grans com a variants estructurals equilibrades i desequilibrades.
No obstant això, calen més estudis per comprendre plenament les limitacions de la WGS i com interpretar un resultat normal. En un futur, la seqüenciació completa del genoma es podria utilitzar com a prova única per detectar la gran majoria d’alteracions genètiques.
Què indica la cobertura?
La intensitat de la cobertura (depth of coverage) indica el nombre de vegades que una base del genoma ha estat seqüenciada. Com més gran és la cobertura més gran és la fiabilitat del mètode. És a dir, es redueix el percentatge de falsos negatius i falsos positius, i és possible fins i tot detectar variants presents en mosaïcisme.
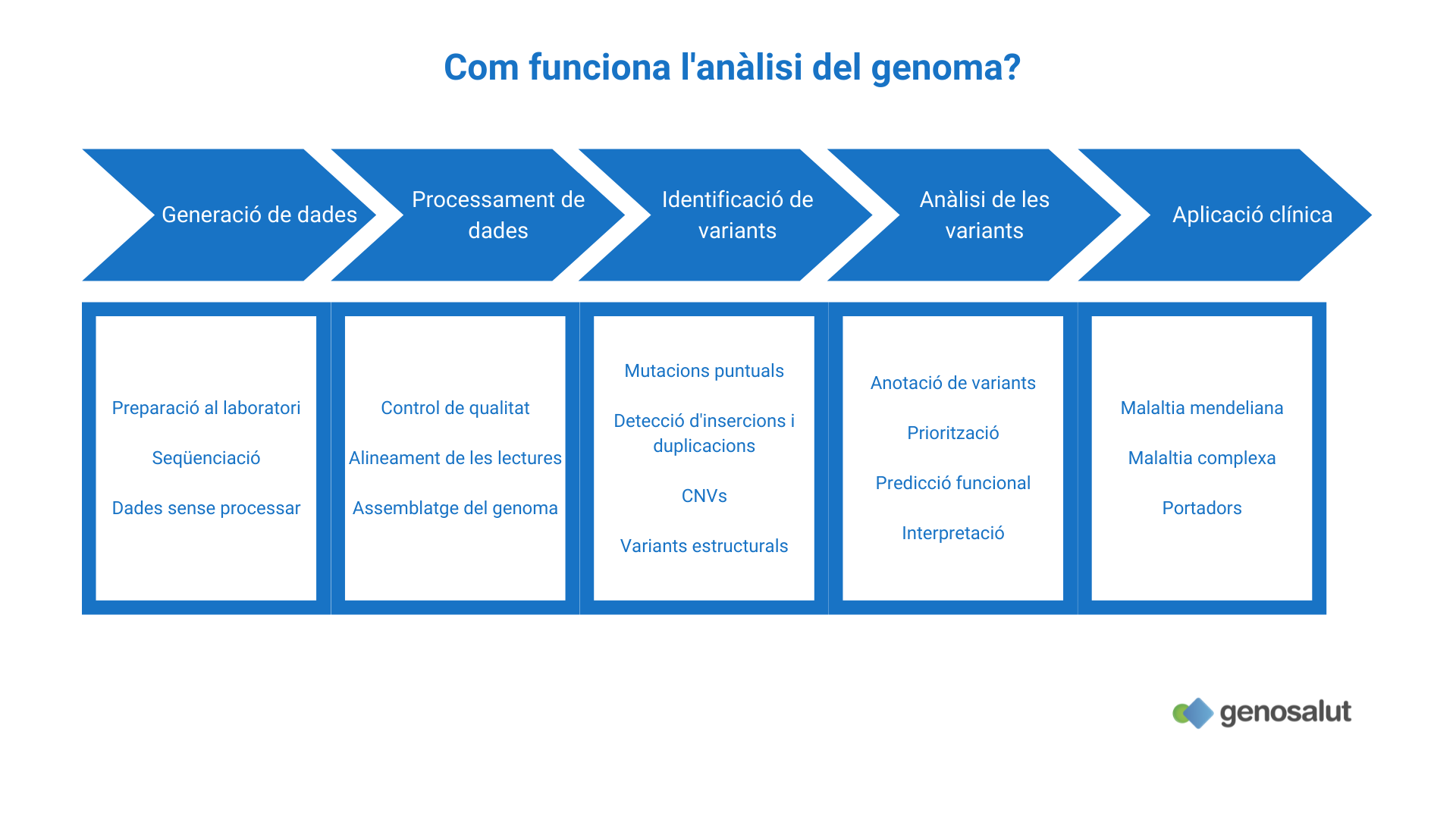
Com funciona la seqüenciació del genoma complet?
La seqüenciació del genoma complet s’engloba dins de les tècniques de seqüenciació massiva o de nova generació. Aquestes tècniques es van començar a desenvolupar a principis de segle amb un nou tipus de seqüenciadors el funcionament dels quals era similar al dels microarrays. A partir de l’ADN d’interès es generaven seqüències curtes (35-500 pb) que eren immobilitzades en un suport sòlid per després ser seqüenciades. Des de llavors, la tecnologia ha evolucionat, és més ràpida, eficaç, fiable i barata. Tot i que s’han desenvolupat diferents plataformes amb diferents detalls tècnics (suport, mètode de seqüenciació i detecció de les seqüències), totes comparteixen algunes característiques comunes que es descriuen a continuació:
- Preparació de les anomenades “llibreries” mitjançant la fragmentació aleatòria de l’ADN (mitjançant sonicació o per mètodes enzimàtics). A continuació, els fragments generats s’uneixen (mitjançant ADN lligasa) a unes molècules anomenades adaptadors. Aquests adaptadors són petites molècules d’ADN que permeten que els fragments generats s’uneixin a seqüències complementàries per a la seva posterior amplificació.
- Amplificació dels fragments per generar amplicons agrupats clonalment (colònies de PCR in situ o polonies, PCR en emulsió, PCR de pont) perquè serveixin com a elements de seqüenciació. D’aquesta manera, els amplicons de la PCR derivats d’una sola molècula de la llibreria acaben agrupats espacialment. Aquest fet és important ja que permet que el senyal lluminós (en incorporar els nucleòtids marcats amb fluorescència a cadascun dels cicles de seqüenciació) sigui prou fort com per a una detecció fiable per part de les càmeres del seqüenciador.
- Seqüenciació: el mètode emprat (entre ells la pirosecuenciació, la seqüenciació per síntesi i la seqüenciació per terminació reversible) depèn de la plataforma que es faci servir. La seqüenciació i detecció de les bases ocorren alhora a totes les molècules d’ADN (seqüenciació massiva i paral·lela).
Les tècniques aquí descrites corresponen a la seqüenciació de segona generació. També hi ha un altre grup de tècniques conegudes com a seqüenciació de tercera generació que utilitzen la seqüenciació d’una sola molècula i la seqüenciació única en temps real, eliminant la necessitat de l’amplificació clona