What is whole genome sequencing?
It is a massive sequencing technique (NGS, next generation sequencing) that allows millions of DNA fragments to be sequenced in parallel, covering the entire genome (WGS, whole genome sequencing)
Secuenciación masiva: análisis del genoma
Whole genome sequencing (WGS) addresses (almost) all of a person’s DNA, both coding (genes) and non-coding regions, and is able to identify most changes in DNA.
This tool, initially exclusive to the research field, has now been introduced into clinical practice for almost a decade. Moreover, it is expected to take on a greater role in healthcare in the future due to the rapidly decreasing costs of sequencing, the ability to produce large volumes of data with today’s sequencers, and the advancing knowledge of genetic variants.
In the future of personalised medicine, whole genome sequence data can be an important tool to guide patient prevention, screening and therapy.
Advantages and disadvantages of whole genome analysis
Advantages of whole genome sequencing
The advantages of this technique are:
- It provides a high-resolution, base-by-base view of the genome.
- It captures both large and small variants that might be missed by targeted approaches.
- Detects single nucleotide variants.
- Detects insertions and deletions (such as the deletion causing spinal muscular atrophy).
- Detects copy number changes.
- Detects large structural variants.
- Detects balanced translocations.
- Detects repeats, e.g. those common in various types of neuromuscular diseases. For example, those known as “polyglutamine” diseases which include, among others, Kennedy’s disease or progressive spinobulbar muscular atrophy, Huntington’s disease, various spinocerebellar ataxias and dentatorubro-pallidoluisian atrophy.
- The significance of variants whose influence on health is currently unknown may be known in the future. Since the data can be retained, the person can be informed in such a case.
- Provides all pharmacogenetic information.
Disadvantages of whole genome sequencing
The greatest difficulty associated with whole genome sequencing is the enormous mass of information it provides, which must be analysed and evaluated to determine what is important and what is not.
Despite growing knowledge in genomics, the functions of many genes are still undetermined and the role of many variants is unknown (it is not known whether they are benign or pathogenic). However, new discoveries may provide valuable information in the future.
Aplicaciones clínicas del análisis del genoma completo
Whole genome analysis covers multiple aspects of health:
- Identifying monogenic diseases (such as thalassaemia).
- Identification of the possible cause in rare diseases.
- Identifying a predisposition to develop a polygenic disease (such as type 2 diabetes mellitus).
- Identifying genetic carriers of recessive diseases, such as cystic fibrosis.
The importance of good data interpretation
Exome sequencing allows the identification of many more variants (point mutations, insertions and deletions) than other techniques, although the significance of some of this information is unknown. Since not all genetic changes affect health, it is sometimes difficult to know whether the variants identified are involved in the health of the person or disease of interest. Sometimes an identified variant is associated with a different genetic disorder that has not yet been diagnosed (called incidental or secondary findings).
This is why it is essential to have a team of experts capable of interpreting the data and answering the questions of doctors and patients.
What genetic variants can be detected by genome sequencing?
The applications of whole genome sequencing (WGS) have been increasing since its inception. While initially used mainly for the detection of single nucleotide changes (SNV, single nucleotide variant or SNP, single nucleotide polymorphism) or point mutations, other types of genetic abnormalities can now be detected with this technique:
- Small copy number variations, such as small insertions and deletions
- Large copy number variations or CNVs (copy number variants)
- Short tandem repeat (STR) sequences
- Runs of homozygosity (ROH) sequences
- Mutations present in a very small percentage of cells (mosaicisms and/or contaminations)
- Major chromosomal rearrangements such as balanced and unbalanced structural variants.
However, further studies are needed to fully understand the limitations of WGS and how to interpret a normal result. In the future, whole genome sequencing could be used as a single test to detect the vast majority of genetic alterations.
What does coverage indicate?
The depth of coverage indicates the number of times a genome base has been sequenced. The higher the coverage, the higher the reliability of the method. In other words, the percentage of false negatives and false positives is reduced, and it is even possible to detect variants present in mosaicism.
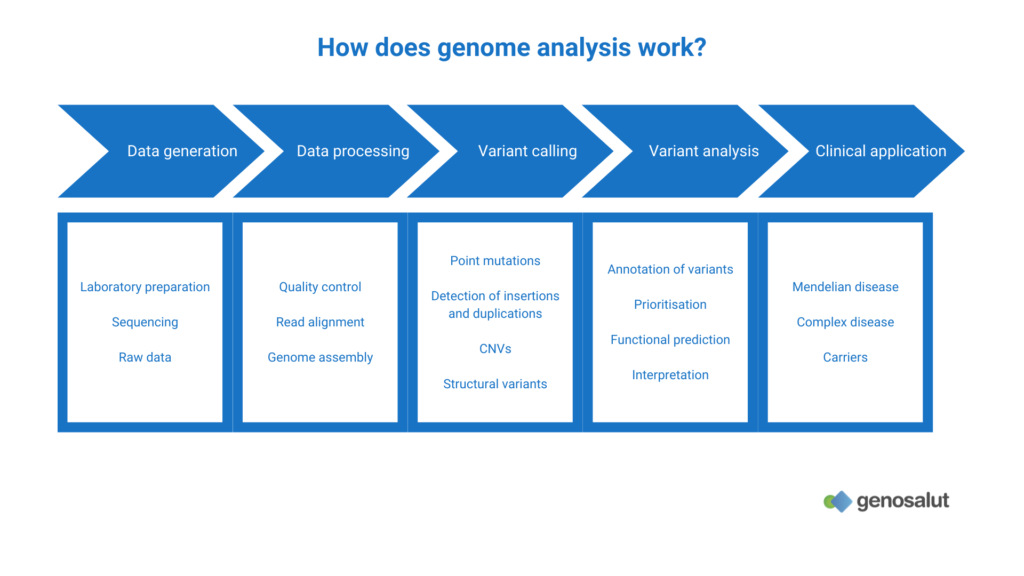
How does whole genome sequencing work?
Whole genome sequencing is one of the massive or next-generation sequencing techniques. These techniques began to be developed at the beginning of the century with a new type of sequencer whose operation was similar to that of microarrays. Short sequences (35-500 bp) were generated from the DNA of interest, immobilised on a solid support and then sequenced. Since then, the technology has evolved, becoming faster, more efficient, reliable and cheaper. Although different platforms have been developed with different technical details (support, sequencing method and sequence detection), they all share some common features which are described below:
- Preparation of the so-called “libraries” by random fragmentation of the DNA (by sonication or enzymatic methods). The fragments generated are then attached (by DNA ligase) to molecules called adapters. These adapters are in turn small DNA molecules that allow the generated fragments to bind to complementary sequences for further amplification.
- Amplification of the fragments to generate clonally pooled amplicons (in situ PCR colonies or polonies, emulsion PCR, bridging PCR) to serve as sequencing elements. In this way, PCR amplicons derived from a single molecule in the library end up spatially clustered. This is important as it allows the light signal (by incorporating the fluorescently labelled nucleotides in each of the sequencing runs) to be strong enough for reliable detection by the sequencer cameras.
- Sequencing: the method employed (including pyrosequencing, sequencing by synthesis and reverse termination sequencing) depends on the platform used. Sequencing and base detection occur at the same time on all DNA molecules (massively parallel sequencing).
The techniques described here correspond to second generation sequencing. There is also another group of techniques known as third generation sequencing that use single molecule sequencing and single real-time sequencing, eliminating the need for clonal amplification.