Was ist die Ganz-Genom-Sequenzierung?
Es handelt sich um eine Hochdurchsatz-Sequenzierungstechnik (NGS, next generation sequencing), mit der Millionen von DNA-Fragmenten parallel sequenziert werden können, die das gesamte Genom abdecken (WGS, whole genome sequencing)
Hochdurchsat-Sequenzierung: Genom-Analyse
Die Sequenzierung des kompletten Genoms (WGS) befasst sich mit (fast) der gesamten DNA einer Person, sowohl mit kodierenden (Genen) als auch mit nicht kodierenden Regionen, und ist in der Lage, die meisten Veränderungen in der DNA zu identifizieren.
Dieses Instrument, das zunächst ausschließlich in der Forschung eingesetzt wurde, wird nun seit fast zehn Jahren im klinischen Alltag verwendet. Außerdem wird erwartet, dass diese Technologie in Zukunft eine größere Rolle im Gesundheitswesen spielen wird, da die Kosten für die Sequenzierung rasch sinken, große Datenmengen erzeugt werden können mit den heutigen Sequenzern und das Wissen über genetische Varianten wächst.
In der Zukunft der personalisierten Medizin können Daten aus dem gesamten Genom ein wichtiges Instrument sein, um Patienten bei Prävention, Screening und Therapie zu unterstützen.
Vorteile und Nachteile der Ganz-Genom-Analyse
Vorteile der Sequenzierung des kompletten Genoms
Die Vorteile dieser Technik sind:
- Sie bietet eine hochauflösende, basengenaue Ansicht des Genoms.
- Sie erfasst sowohl große als auch kleine Varianten, die bei gezielten Ansätzen möglicherweise übersehen werden.
- Detektiert einzelne Nukleotidvarianten.
- Erkennt Insertionen und Deletionen (wie die Deletion, die spinale Muskelatrophie verursacht).
- Erkennt Änderungen der Kopienzahl.
- Entdeckt große strukturelle Varianten.
- Erkennt balancierte Translokationen.
- Erkennt Wiederholungen, die z. B. bei verschiedenen neuromuskulären Erkrankungen auftreten. Zum Beispiel die so genannten „Polyglutamin“-Krankheiten, zu denen unter anderem die Kennedy-Krankheit oder die progressive spinobulbäre Muskelatrophie, die Huntington-Krankheit, verschiedene spinozerebellare Ataxien und die dentatorubro-pallidoluisische Atrophie gehören.
- Die Bedeutung von Varianten, deren Einfluss auf die Gesundheit derzeit noch unbekannt ist, könnte in Zukunft bekannt werden. Da die Daten aufbewahrt werden können, kann die Person in einem solchen Fall informiert werden.
- Sie enthält alle pharmakogenetischen Informationen.
Nachteile der Sequenzierung des kompletten Genoms
Die größte Schwierigkeit bei der Sequenzierung des gesamten Genoms besteht in der enormen Menge an Informationen, die analysiert und bewertet werden müssen, um festzustellen, was wichtig ist und was nicht.
Trotz des zunehmenden Wissens in der Genomik sind die Funktionen vieler Gene noch immer unklar und die Rolle vieler Varianten ist unbekannt (es ist nicht bekannt, ob sie gutartig oder pathogen sind). Neue Entdeckungen könnten jedoch in Zukunft wertvolle Informationen liefern.
Klinische Anwendungen der Ganz-Genom-Analyse
Die Analyse des gesamten Genoms deckt zahlreiche Aspekte der Gesundheit ab:
- Identifizierung monogener Krankheiten (z. B. Thalassämie).
- Identifizierung der möglichen Ursache bei seltenen Krankheiten.
- Identifizierung einer Veranlagung zur Entwicklung einer polygenen Krankheit (z. B. Diabetes mellitus Typ 2).
- Identifizierung von Genträgern rezessiver Krankheiten, wie z. B. Mukoviszidose.
La importancia de una buena interpretación de los datos
Die Genom-Sequenzierung ermöglicht die Identifizierung von viel mehr Varianten (Punktmutationen, Insertionen und Deletionen) als andere Techniken, obwohl die Bedeutung einiger dieser Informationen unbekannt ist. Da sich nicht alle genetischen Veränderungen auf die Gesundheit auswirken, ist es manchmal schwierig festzustellen, ob die identifizierten Varianten mit der Gesundheit der betreffenden Person oder der betreffenden Krankheit zusammenhängen. Manchmal wird eine identifizierte Variante mit einer anderen genetischen Störung in Verbindung gebracht, die noch nicht diagnostiziert wurde (sogenannte Zufalls- oder Sekundärbefunde).
Deshalb ist es wichtig, über ein Team von Experten zu verfügen, das in der Lage ist, die Daten zu interpretieren und die Fragen der Ärzte und Patienten zu beantworten.
Welche genetischen Varianten kann die Genom-Sequenzierung nachweisen?
Die Anwendungen der Sequenzierung ganzer Genome (whole genome sequencing, WGS) haben seit ihren Anfängen zugenommen. Ursprünglich wurde diese Technik hauptsächlich für den Nachweis von Einzelnukleotidveränderungen (SNV, single nucleotide variant oder SNP, single nucleotide polymorphism) oder Punktmutationen verwendet, doch inzwischen können auch andere Arten von genetischen Anomalien mit dieser Technik nachgewiesen werden:
- Kleine Kopienzahlvariationen, wie kleine Insertionen und Deletionen
- Große Kopienzahlvariationen oder CNVs (Kopienzahlvarianten)
- Kurztandem-Repeat-Sequenzen (STR)
- Runs of Homozygosity (ROH)-Sequenzen
- Mutationen in einem sehr geringen Prozentsatz der Zellen (Mosaikismen und/oder Kontaminationen)
- Größere chromosomale Umlagerungen wie balancierte und unbalancierte Strukturvarianten.
Es sind jedoch weitere Studien erforderlich, um die Grenzen von WGS vollständig zu verstehen und um zu wissen, wie ein normales Ergebnis zu interpretieren ist. Künftig könnte die Sequenzierung des gesamten Genoms als einziger Test verwendet werden, um die große Mehrheit der genetischen Veränderungen nachzuweisen.
Was besagt die Abdeckung?
Die Tiefe der Abdeckung (depth of coverage) gibt an, wie oft eine Genombasis sequenziert wurde. Je höher der Erfassungsgrad, desto zuverlässiger ist die Methode. Mit anderen Worten: Der Prozentsatz falsch negativer und falsch positiver Ergebnisse wird reduziert, und es ist sogar möglich, Varianten zu erkennen, die in Mosaik vorkommen.
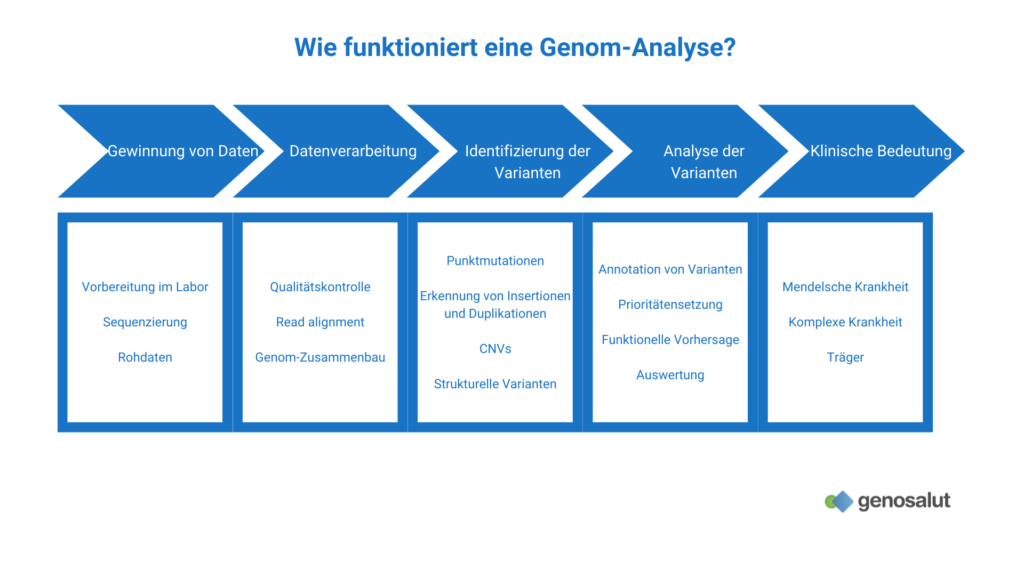
Wie funktioniert die Ganz-Genom-Sequenzierung?
Die Sequenzierung des gesamten Genoms gehört zu den Hochdurchsatz oder next-generation-Sequenzierungstechniken. Die Entwicklung dieser Techniken begann zu Beginn des Jahrhunderts mit einer neuen Art von Sequenzer, dessen Funktionsweise der von Microarrays ähnelte. Kurze Sequenzen (35-500 bp) wurden aus der interessierenden DNA erzeugt, auf einem festen Träger immobilisiert und dann sequenziert. Seitdem hat sich die Technologie weiterentwickelt und ist schneller, effizienter, zuverlässiger und billiger geworden. Obwohl verschiedene Plattformen mit unterschiedlichen technischen Details (Unterstützung, Sequenzierungsmethode und Sequenzerkennung) entwickelt wurden, haben sie alle einige gemeinsame Merkmale, die im Folgenden beschrieben werden:
- Herstellung der so genannten „Bibliotheken“ durch zufällige Fragmentierung der DNA (durch Beschallung oder enzymatische Methoden). Die entstandenen Fragmente werden dann (durch DNA-Ligase) an sogenannte Adaptermoleküle angehängt. Bei diesen Adaptern handelt es sich wiederum um kleine DNA-Moleküle, die es den erzeugten Fragmenten ermöglichen, an komplementäre Sequenzen zu binden, um sie weiter zu amplifizieren.
- Amplifikation der Fragmente zur Erzeugung klonal gepoolter Amplikons (in situ PCR-Kolonien oder Polonien, Emulsions-PCR, Bridging PCR), die als Sequenzierungselemente dienen. Auf diese Weise werden PCR-Amplikone, die von einem einzigen Molekül in der Bibliothek stammen, räumlich geclustert. Dies ist wichtig, damit das Lichtsignal (durch die Einbeziehung der fluoreszenzmarkierten Nukleotide in jeden Sequenzierungslauf) stark genug ist, um von den Kameras des Sequenzers zuverlässig erkannt zu werden.
- Sequenzierung: Die verwendete Methode (einschließlich Pyrosequenzierung, Sequenzierung durch Synthese und Reverse-Termination-Sequenzierung) hängt von der verwendeten Plattform ab. Sequenzierung und Basendetektion finden bei allen Molekülen gleichzeitig statt.
Die hier beschriebenen Techniken entsprechen der Sequenzierung der zweiten Generation. Es gibt auch eine andere Gruppe von Techniken, die als Sequenzierung der dritten Generation bekannt sind und die Einzelmolekülsequenzierung und Einzel-Echtzeitsequenzierung verwenden, wodurch die Notwendigkeit einer klonalen Amplifikation entfällt.