¿Qué es la secuenciación del genoma completo?
Es una técnica de secuenciación masiva (NGS, next generation sequencing) que permite secuenciar, de forma paralela, millones de fragmentos de ADN, que cubren todo el genoma (WGS, whole genome sequencing)
Secuenciación masiva: análisis del genoma
La secuenciación del genoma completo (WGS) aborda (casi) todo el ADN de la persona, tanto las regiones codificantes (genes) como no codificantes, y es capaz de identificar la mayoría de los cambios en el ADN.
Esta herramienta, al principio exclusiva del campo de la investigación, lleva ya casi una década introducida en la práctica clínica. Además, se espera que en el futuro adquiera un papel más relevante dentro del ámbito sanitario gracias al rápido descenso de los costes de secuenciación, la capacidad de producir grandes volúmenes de datos con los secuenciadores actuales y el avance del conocimiento de las variantes genéticas.
En el futuro de la medicina personalizada, los datos de la secuencia del genoma completo pueden ser una herramienta importante para guiar la prevención, detección y terapia de los pacientes.
Ventajas y desventajas del análisis del genoma completo
Ventajas de la secuenciación del genoma completo
Las ventajas de esta técnica son:
- Proporciona una visión de alta resolución, base por base, del genoma.
- Captura tanto las variantes grandes como las pequeñas que podrían pasar desapercibidas con enfoques específicos.
- Detecta variantes de un solo nucleótido.
- Detecta inserciones y deleciones (como la deleción causante de la atrofia muscular espinal).
- Detecta cambios en el número de copias.
- Detecta grandes variantes estructurales.
- Detecta translocaciones equilibradas.
- Detecta repeticiones, por ejemplo aquellas comunes en diversos tipos de enfermedades neuromusculares. Por ejemplo, aquellas conocidas como enfermedades «poliglutamínicas» que incluyen entre otras, la enfermedad de Kennedy o atrofia muscular progresiva espinobulbar, la enfermedad de Huntington, varias ataxias espinocerebelosas y la atrofia dentatorubro-pálidoluisiana.
- El significado de variantes cuya influencia sobre la salud se desconoce en la actualidad puede conocerse en el futuro. Dado que los datos se pueden conservar, la persona puede ser informada en tal caso.
- Proporciona toda la información farmacogenética.
Desventajas de la secuenciación del genoma completo
La mayor dificultad asociada a la secuenciación del genoma completo es la enorme masa de información que proporciona, que debe ser analizada y evaluada para determinar lo que es importante o lo que no lo es.
A pesar de que los conocimientos en genómica son cada vez mayores, las funciones de muchos genes son todavía indeterminadas y se desconoce el papel de muchas variantes (no se sabe si son benignas o patógenas). No obstante, gracias a nuevos descubrimientos, puede ser una información valiosa en el futuro.
Aplicaciones clínicas del análisis del genoma completo
El análisis del genoma completo abarca múltiples aspectos de la salud:
- Identificar enfermedades monogénicas (como la talasemia).
- Identificación de la posible causa en enfermedades raras.
- Identificar una predisposición a desarrollar una enfermedad poligénica (como la diabetes mellitus tipo 2).
- Identificar a los portadores genéticos de enfermedades recesivas, como la fibrosis quística.
La importancia de una buena interpretación de los datos
La secuenciación del genoma permite la identificación de muchas más variantes que otras técnicas, no obstante se desconoce el significado de parte de esta información. Dado que no todos los cambios genéticos afectan a la salud, en ocasiones es difícil saber si las variantes identificadas están implicadas en la salud de la persona o la enfermedad de interés. A veces, una variante identificada se asocia con un trastorno genético diferente que aún no se ha diagnosticado (lo que se denomina hallazgos incidentales o secundarios).
Por ello es imprescindible contar con un equipo de profesionales expertos capaces de interpretar los datos y dar respuesta a las preguntas de los médicos y los pacientes.
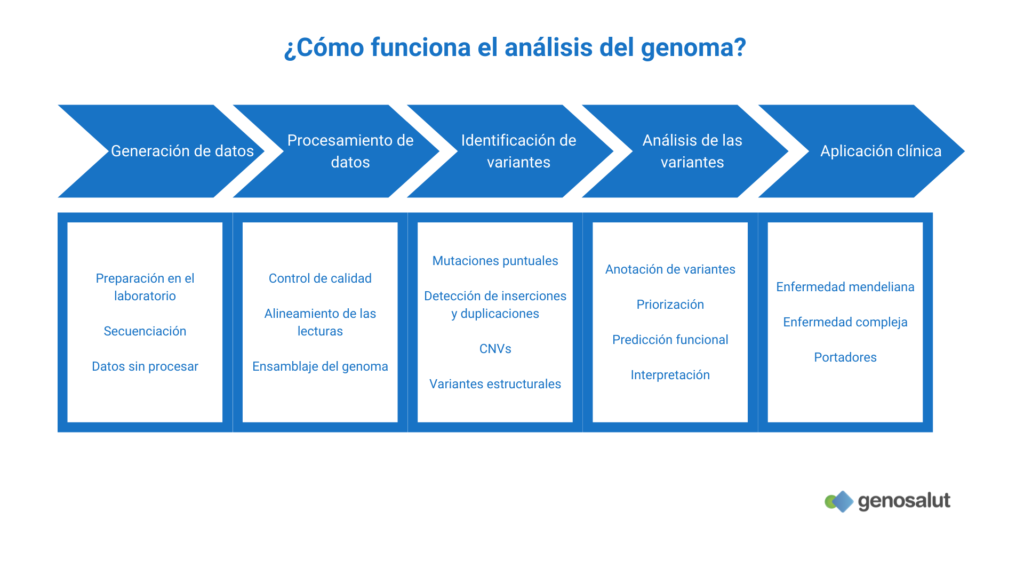
¿Qué variantes genéticas se pueden detectar mediante la secuenciación del genoma?
Las aplicaciones de la secuenciación del genoma completo (WGS, whole genome sequencing) han ido aumentando desde sus inicios. Si bien al principio se usaba principalmente para la detección de cambios en un sólo nucleótido (SNV, single nucleotide variant o SNP, single nucleotide polymorphism) o mutaciones puntuales, actualmente con esta técnica ya se pueden detectar otro tipo de anomalías genéticas entre las que se encuentran:
- Pequeñas variaciones en el número de copias tales como pequeñas inserciones y deleciones
- Grandes variaciones en el número de copias o CNVs (copy number variants)
- Repeticiones cortas en tándem (STR, short tandem repeat)
- Secuencias de homocigosidad (ROH, runs of homozygosity)
- Mutaciones presentes en un porcentaje muy pequeño de células (mosaicismos y/o contaminaciones)
- Reordenamientos cromosómicos mayores como variantes estructurales equilibradas y desequilibradas.
No obstante, son necesarios más estudios para comprender plenamente las limitaciones de la WGS y cómo interpretar un resultado normal. En un futuro, la secuenciación completa del genoma podría ser utilizada como prueba única para detectar la gran mayoría de alteraciones genéticas.
¿Qué indica la cobertura?
La intensidad de la cobertura (depth of coverage) indica número de veces que una base del genoma ha sido secuenciada. Cuanto mayor es la cobertura mayor es la fiabilidad del método. Es decir, se reduce el porcentaje de falsos negativos y falsos positivos, y es posible incluso detectar variantes presentes en mosaicismo.
¿Cómo funciona la secuenciación del genoma completo?
La secuenciación del genoma completo se engloba dentro de las técnicas de secuenciación masiva o de nueva generación. Estas técnicas se empezaron a desarrollar a principios de siglo con un nuevo tipo de secuenciadores cuyo funcionamiento era similar al de los microarrays. A partir del ADN de interés se generaban secuencias cortas (35-500 pb) que eran inmovilizadas en un soporte sólido para luego ser secuenciadas. Desde entonces, la tecnología ha evolucionado, es más rápida, eficaz, fiable y barata. Aunque se han desarrollado diferentes plataformas con distintos detalles técnicos (soporte, método de secuenciación y detección de las secuencias), todas comparten algunas características comunes que se describen a continuación:
- Preparación de las llamadas «librerías» mediante la fragmentación aleatoria del ADN (mediante sonicación o por métodos enzimáticos). A continuación, los fragmentos generados se unen (mediante ADN ligasa) a unas moléculas llamadas adaptadores. Estos adaptadores son a su vez pequeñas moléculas de ADN que permiten que los fragmentos generados se unan a secuencias complementarias para su posterior amplificación.
- Amplificación de los fragmentos para generar amplicones agrupados clonalmente (colonias de PCR in situ o polonies, PCR en emulsión, PCR de puente) para que sirvan como elementos de secuenciación. De esta forma, los amplicones de la PCR derivados de una sola molécula de la librería terminan agrupados espacialmente. Este hecho es importante ya que permite que la señal luminosa (al incorporar los nucleótidos marcados con fluorescencia en cada uno de los ciclos de secuenciación) sea lo suficientemente fuerte como para una detección fiable por parte de las cámaras del secuenciador.
- Secuenciación: el método empleado (entre ellos la pirosecuenciación, la secuenciación por síntesis y la secuenciación por terminación reversible) depende de la plataforma que se utilice. La secuenciación y detección de las bases ocurren al mismo tiempo en todas las moléculas de ADN (secuenciación masiva y paralela).
Las técnicas aquí descritas se corresponden a la secuenciación de segunda generación. Existe también otro grupo de técnicas conocidas como secuenciación de tercera generación que utilizan la secuenciación de una sola molécula y la secuenciación única en tiempo real, eliminando la necesidad de la amplificación clonal.