Andrade's disease or familial amyloid polyneuropathy
Analysis of mutations associated with Andrade disease or familial amyloid polyneuropathy (FAP).
Price
150€ (includes genetic counselling)
Time to result
3 weeks
Laboratorio diagnóstico genético
Analysis of mutations associated with Andrade disease or familial amyloid polyneuropathy (FAP).
Price
150€ (includes genetic counselling)
Time to result
3 weeks
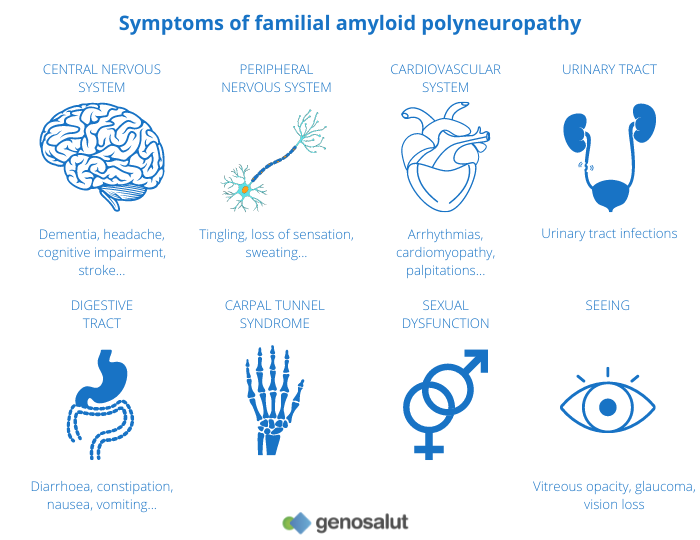
Andrade’s disease or familial amyloid polyneuropathy (FAP), also known as transthyretin amyloid polyneuropathy. It is a progressive autonomic sensory-motor neuropathy that appears in adulthood.
The first manifestations are paraesthesia, pain or trophic lesions of the feet, gastrointestinal disorders or weight loss. The most pronounced sensory loss involves pain and thermal sensation. Motor loss occurs later. Autonomic features include postural hypotension and gastrointestinal and genitourinary disorders. Cardiac involvement is common, as are ocular or renal complications.
Although clinically heterogeneous and its clinical presentation depends on genotype and geographic origin, Andrade’s disease is severe and disabling. Death occurs on average about 11 years after the onset of the first symptoms and may occur suddenly or as a result of infections or cachexia.
At Genosalut we analyse the most frequent variant in the Balearic Islands (TTR Val30Met) as well as the other mutations related to this disease.
We also offer personalised advice and genetic counselling to affected families.
In the case of the Mallorcan population, first of all, the presence of the TTR Val30Met variant (NM_000371.4(TTR):c.148G>A (p.Val50Met)
TTR Val30Met, TTR V30M, rs28933979) by sequencing or real-time PCR.
We also analyse the approx. 40 mutations associated with this disease by sequencing (exons 1, 2, 3 and 4) or exome analysis.
Experience
At Genosalut, we have more than 10 years of experience in counselling people with conditions where a genetic cause has been identified or is thought to be possible.
Proximity
We are a close laboratory, we respond personally and we take the time to explain the report in detail to doctors and patients.
Professional interpretation of results
Because of our knowledge and experience, we are able to accurately interpret genetic results and offer professional advice.
Reference in the field
We are the point of contact for patients, doctors and clinics in all areas of human genetic diagnostics and prevention.
Andrade disease is transmitted as an autosomal dominant trait of variable penetrance and is caused by mutations in the TTR gene.
More than 40 TTR mutations have been identified so far, associated with varying patterns of organ involvement, age of onset and disease progression.
The most common variant is the TTR Val30Met substitution (rs28933979), for which several endemic foci have been identified, especially in Portugal, Japan and Sweden. In Spain there are endemic foci in Mallorca and Huelva. The TTR Val30Met phenotype varies between these countries. Protective factors are thought to be involved. In the case of the Mallorcan population, the TTR Gly6Ser polymorphism (rs28933979) could be one of these protective factors. The worldwide prevalence is unknown, although the prevalence among the general population in Japan has recently been estimated at 1:1000000.
You will know if you are a carrier of a mutation associated with Andrade's disease
The detection of amyloid-associated TTR mutations is required for the diagnosis of Andrade disease. However, the identification of a disease-causing mutation is not considered diagnostic, because the genetic penetrance is variable. During the genetic counselling session we clarify all doubts with our patients.
You will know if you can pass on the risk to your offspring
If a mutation is detected, it is possible to pass it on to offspring. Therefore, knowledge can be important for future family planning.
Contact us through the form, by e-mail or by telephone to make an appointment with us.
You can also consult your doctor for information on the possibilities of genetic testing.
In our genetic diagnostics laboratory we analyse the sample with the latest technology.
We provide a detailed description of the results and, if necessary, genetic counselling.
Detection of amyloid-associated TTR mutations is required for diagnosis. Even so, the identification of a disease-causing mutation is not considered diagnostic, because genetic penetrance is variable. Clinical observation and biopsy of tissues (such as nerve, kidney, labial salivary glands, subcutaneous fatty tissue or rectal mucosa) are necessary for a definitive diagnosis: amyloid deposits are detected by Congo red staining by conventional light microscopy and by green birefringence by polarised light microscopy.
At Genosalut we offer genetic counselling to affected families to explain the implications for the patient and their relatives. Some considerations to take into account:
Management of familial amyloid polyneuropathy must be multidisciplinary, involving a neurologist, geneticist, cardiologist and liver surgeon.
Liver transplantation is currently the only treatment to prevent the synthesis of amyloidogenic TTR variants. Liver transplantation can slow the progression of the disease in its early stages.
Symptomatic treatments are essential for autonomic sensorimotor neuropathies with visceral complications.
Opening hours
Monday to Friday from 9.00 am to 1.00 pm
+34 616 59 01 65
info@genosalut.com
Camí dels Reis, 308 (Clínica Palma Planas)
Contact form
First genetic diagnosis laboratory in the Balearic Islands
Professionals with experience in medical genetics
Detailed report of the results
Personalised attention for each patient
Wide range of genetic tests
Cutting-edge technology